
Page 394 - Advanced Organic Chemistry Part A - Structure and Mechanisms, 5th ed (2007) - Carey _ Sundberg
P. 394
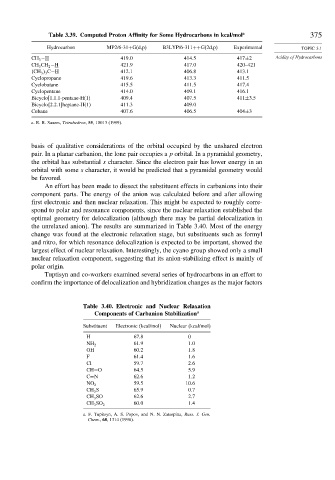
Table 3.39. Computed Proton Affinity for Some Hydrocarbons in kcal/mol a 375
Hydrocarbon MP2/6-31+G(d,p) B3LYP/6-311++G(2d,p) Experimental TOPIC 3.1
CH 3 −H 419 0 414 5 417±2 Acidity of Hydrocarbons
CH 3 CH 2 −H 421 9 417 0 420–421
CH 3 3 C−H 412 1 406 8 413.1
Cyclopropane 419 6 413 3 411.5
Cyclobutane 415 5 411 5 417.4
Cyclopentane 414 0 409 1 416.1
Bicyclo[1.1.1-pentane-H(1) 409 4 407 5 411±3.5
Bicyclo{2.2.1]heptane-H(1) 411 3 409 0
Cubane 407 6 406 5 404±3
a. R. R. Sauers, Tetrahedron, 55, 10013 (1999).
basis of qualitative considerations of the orbital occupied by the unshared electron
pair. In a planar carbanion, the lone pair occupies a p orbital. In a pyramidal geometry,
the orbital has substantial s character. Since the electron pair has lower energy in an
orbital with some s character, it would be predicted that a pyramidal geometry would
be favored.
An effort has been made to dissect the substituent effects in carbanions into their
component parts. The energy of the anion was calculated before and after allowing
first electronic and then nuclear relaxation. This might be expected to roughly corre-
spond to polar and resonance components, since the nuclear relaxation established the
optimal geometry for delocalization (although there may be partial delocalization in
the unrelaxed anion). The results are summarized in Table 3.40. Most of the energy
change was found at the electronic relaxation stage, but substituents such as formyl
and nitro, for which resonance delocalization is expected to be important, showed the
largest effect of nuclear relaxation. Interestingly, the cyano group showed only a small
nuclear relaxation component, suggesting that its anion-stabilizing effect is mainly of
polar origin.
Tuptisyn and co-workers examined several series of hydrocarbons in an effort to
confirm the importance of delocalization and hybridization changes as the major factors
Table 3.40. Electronic and Nuclear Relaxation
Components of Carbanion Stabilization a
Substituent Electronic (kcal/mol) Nuclear (kcal/mol)
H 67 8 0
61 9 1 0
NH 2
OH 60 2 1 8
F 61 4 1 6
Cl 59 7 2 6
CH=O 64 5 5 9
C≡N 62 6 1 2
59 5 10 6
NO 2
CH 3 S 65 9 0 7
CH 3 SO 62 6 2 7
60 0 1 4
CH 3 SO 2
a. F. Tupitsyn, A. S. Popov, and N. N. Zatsepina, Russ. J. Gen.
Chem., 68, 1314 (1998).