
Page 356 - Fundamentals of Gas Shale Reservoirs
P. 356
336 MOLECULAR SIMULATION OF GAS ADSORPTION IN MINERALS AND COAL
sample to the other (Day et al., 2008; Ottiger et al., 2008; 1.4
St George and Barakat, 2001).
The system simulated consists of coal and pure com 1.2
ponent of CH . The initial configuration consists of 12
4
randomly placed coal molecules. All molecules are randomly 1.0
placed in an empty space of a simulation box, which is
large enough to accommodate the coal molecules and has Absolute CH 4 adsorption (mmol/g) 0.8
3
x, y, and z‐dimensions of 3.2 × 3.2 × 3.2 nm , shown in 0.6
Figure 15.9. Simulation (0 wt% water)
In MD simulations, the behavior of a large number of 0.4 Simulation (1.2 wt% water)
atoms in the simulation box is followed as a function of Simulation (3.0 wt% water)
Langmuir t (0 wt% water)
time by Newton’s laws. Periodic boundary conditions are 0.2 Langmuir t (1.2 wt% water)
applied in three directions. The movement of the atoms Langmuir t (3.0 wt% water)
within the simulation box is governed by an interatomic 0.0
interaction potential between all atoms (force field). This 0 2 4 6 8 10
is the core of the MD simulations, as the accuracy of P (MPa)
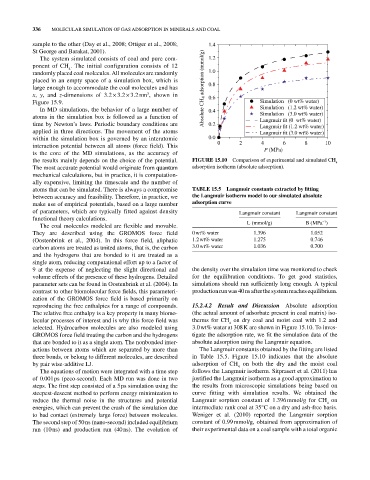
the results mainly depends on the choice of the potential. FIGURE 15.10 Comparison of experimental and simulated CH
4
The most accurate potential would originate from quantum adsorption isotherm (absolute adsorption).
mechanical calcu lations, but in practice, it is computation
ally expensive, limiting the timescale and the number of
atoms that can be simulated. There is always a compromise TAbLE 15.5 Langmuir constants extracted by fitting
between accuracy and feasibility. Therefore, in practice, we the Langmuir isotherm model to our simulated absolute
make use of empirical potentials, based on a large number adsorption curve
of parameters, which are typically fitted against density Langmuir constant Langmuir constant
functional theory calculations. −1
The coal molecules modeled are flexible and movable. L (mmol/g) B (MPa )
They are described using the GROMOS force field 0 wt% water 1.396 1.052
(Oostenbrink et al., 2004). In this force field, aliphatic 1.2 wt% water 1.275 0.746
carbon atoms are treated as united atoms, that is, the carbon 3.0 wt% water 1.036 0.700
and the hydrogens that are bonded to it are treated as a
single atom, reducing computational effort up to a factor of
9 at the expense of neglecting the slight directional and the density over the simulation time was monitored to check
volume effects of the presence of these hydrogens. Detailed for the equilibration conditions. To get good statistics,
parameter sets can be found in Oostenbrink et al. (2004). In simulations should run sufficiently long enough. A typical
contrast to other biomolecular force fields, this parameteri production run was 40 ns after the system reaches equilibrium.
zation of the GROMOS force field is based primarily on
reproducing the free enthalpies for a range of compounds. 15.2.4.2 Result and Discussion Absolute adsorption
The relative free enthalpy is a key property in many biomo (the actual amount of adsorbate present in coal matrix) iso
lecular processes of interest and is why this force field was therms for CH on dry coal and moist coal with 1.2 and
4
selected. Hydrocarbon molecules are also modeled using 3.0 wt% water at 308 K are shown in Figure 15.10. To inves
GROMOS force field treating the carbon and the hydrogens tigate the adsorption rate, we fit the simulation data of the
that are bonded to it as a single atom. The nonbonded inter absolute adsorption using the Langmuir equation.
actions between atoms which are separated by more than The Langmuir constants obtained by the fitting are listed
three bonds, or belong to different molecules, are described in Table 15.5. Figure 15.10 indicates that the absolute
by pair wise‐additive LJ. adsorption of CH on both the dry and the moist coal
4
The equations of motion were integrated with a time step follows the Langmuir isotherm. Sitprasert et al. (2011) has
of 0.001 ps (peco‐second). Each MD run was done in two justified the Langmuir isotherm as a good approximation to
steps. The first step consisted of a 5 ps simulation using the the results from microscopic simulations being based on
steepest‐descent method to perform energy minimization to curve fitting with simulation results. We obtained the
reduce the thermal noise in the structures and potential Langmuir sorption constant of 1.396 mmol/g for CH on
4
energies, which can prevent the crash of the simulation due intermediate rank coal at 35°C on a dry and ash‐free basis.
to bad contact (extremely large force) between molecules. Weniger et al. (2010) reported the Langmuir sorption
The second step of 50 ns (nano‐second) included equilibrium constant of 0.99 mmol/g, obtained from approximation of
run (10 ns) and production run (40 ns). The evolution of their experimental data on a coal sample with a total organic