
Page 180 - Macromolecular Crystallography
P. 180
MODEL BUILDING, REFINEMENT, AND VALIDATION 169
density modification as implemented in RESOLVE, system that will judge the model completeness and
’standard’ density modification techniques (e.g. learn from the accumulated history. Although great
Abrahams, 1997) or new approaches (e.g. PIRATE advances have been made in the field of crys-
from the CCP4 suite). RESOLVE and ARP/wARP tallographic model building and refinement, fur-
both have a good chance of bootstrapping a correct ther effort must be invested to turn this technique
model after density modification. into truly accessible and easy-to-use tool for biol-
With poor models or lower resolution is may ogists. The coming years will no doubt see a greater
be advantageous to compute model phases and involvement of researchers with different expertise
then treat them as ’experimental’ ones. Guidelines in achieving this goal.
described in the preceding section would then be A major challenge will be the refinement based on
applicable. multiple diffraction data sets. For example, a protein
with two models available at high resolution and a
complex structure determined at lower resolution,
one could envisage the simultaneous refinement of
11.5 Concluding remarks
all three structures. Areas that are unaffected by the
While the automation of initial model building has formation of the complex and short-range features
largely been alleviated (at least for good quality elec- (i.e. bonds and angles that are likely to be invariant)
tron density maps and sufficient resolution of the would benefit from the high resolution data, while
X-ray data), an increasing number of non-expert long-range features (relative placement of helices for
crystallographer users do need a substantial amount example) could be determined from the contribu-
of time to complete and finalize the model. This tion of the low resolution data. Such developments
step that a few years back seemed to be of sec- still await theoretical underpinning and implemen-
ondary importance is now becoming a bottleneck tations. Similar ideas can be used for the refinement
and will be one of the targets of the new develop- of the same protein in differently liganded states.
ments. Inherent components of this task are building With the continuously increasing usage of X-ray
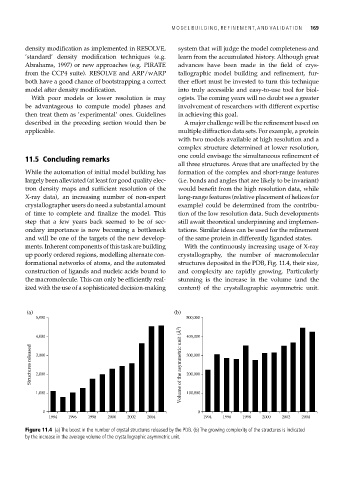
up poorly ordered regions, modelling alternate con- crystallography, the number of macromolecular
formational networks of atoms, and the automated structures deposited in the PDB, Fig. 11.4, their size,
construction of ligands and nucleic acids bound to and complexity are rapidly growing. Particularly
the macromolecule. This can only be efficiently real- stunning is the increase in the volume (and the
ized with the use of a sophisticated decision-making content) of the crystallographic asymmetric unit.
(a) (b)
5,000 500,000
4,000 400,000
Structures released 3,000 Volume of the asymmetric unit (Å 3 ) 300,000
200,000
2,000
1,000 100,000
0 0
1994 1996 1998 2000 2002 2004 1994 1996 1998 2000 2002 2004
Figure 11.4 (a) The boost in the number of crystal structures released by the PDB. (b) The growing complexity of the structures is indicated
by the increase in the average volume of the crystallographic asymmetric unit.