
Page 324 - Handbook Of Multiphase Flow Assurance
P. 324
Computer modeling of interaction between a hydrate surface and an inhibitor 323
4.00
N
O
1 C
3.00
2 3
Distance from oxygen to
Oxygen
Nitrogen
2.00 Carbonyl
carbon
Carbon 1
Carbon 2
Carbon 3
1.00
0.00
0.00 2.00 4.00 6.00 8.00
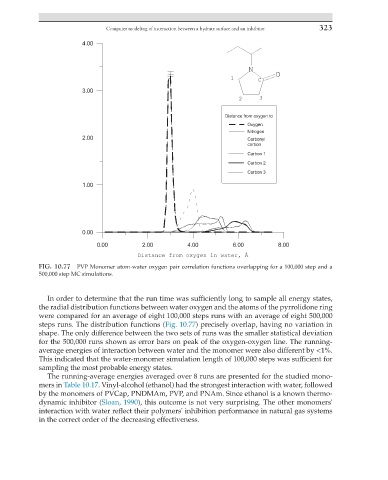
FIG. 10.77 PVP Monomer atom-water oxygen pair correlation functions overlapping for a 100,000 step and a
500,000 step MC simulations.
In order to determine that the run time was sufficiently long to sample all energy states,
the radial distribution functions between water oxygen and the atoms of the pyrrolidone ring
were compared for an average of eight 100,000 steps runs with an average of eight 500,000
steps runs. The distribution functions (Fig. 10.77) precisely overlap, having no variation in
shape. The only difference between the two sets of runs was the smaller statistical deviation
for the 500,000 runs shown as error bars on peak of the oxygen-oxygen line. The running-
average energies of interaction between water and the monomer were also different by <1%.
This indicated that the water-monomer simulation length of 100,000 steps was sufficient for
sampling the most probable energy states.
The running-average energies averaged over 8 runs are presented for the studied mono-
mers in Table 10.17. Vinyl-alcohol (ethanol) had the strongest interaction with water, followed
by the monomers of PVCap, PNDMAm, PVP, and PNAm. Since ethanol is a known thermo-
dynamic inhibitor (Sloan, 1990), this outcome is not very surprising. The other monomers'
interaction with water reflect their polymers' inhibition performance in natural gas systems
in the correct order of the decreasing effectiveness.