
Page 106 - Hydrogeology Principles and Practice
P. 106
HYDC03 12/5/05 5:36 PM Page 89
Chemical hydrogeology 89
3.6.2 Energetic approach to chemical equilibrium
In this approach to chemical equilibrium, the most
stable composition of a mixture of reactants is the
composition having the lowest energy. This more
rigorous thermodynamic treatment, compared with
the kinetic approach, involves enthalpy, the heat
content, H, at constant pressure, and entropy, S, a
measure of the disorder of a system. The change in
−1
enthalpy (∆H, measured in J mol ) in a reaction is a
direct measure of the energy emitted or absorbed.
The change in entropy in most reactions (∆S, meas-
−1
−1
ured in J mol K ) proceeds to increase disorder,
for example by splitting a compound into its con-
stituent ions. For a reversible process, the change in
entropy is equal to the amount of heat taken up by a
reaction divided by the absolute temperature, T, at
which the heat is absorbed.
The total energy released, or the energy change in
going from reactants to products, is termed the Gibbs
−1
free energy, G (measured in kJ mol ). If energy is
released, in which case the products have lower free
energy than the reactants, G is considered negative.
The change in Gibbs free energy is defined as:
∆G =∆H − T∆S eq. 3.8
By convention, elements in their standard state (25°C
and 1 atmosphere pressure) are assigned enthalpy and
free energy values of zero. Standard state thermodyn-
o
amic data, indicated by the superscript , and tabul-
ated as values of standard free energies, enthalpies
and entropies, are given in most geochemistry and
aqueous chemistry textbooks, for example Krauskopf
and Bird (1995) and Stumm and Morgan (1981).
o
Values of ∆G for different reactions can be calculated
by simple arithmetic combination of the tabulated
o
values. Any reaction with a negative ∆G value will,
in theory, proceed spontaneously (the chemical
equivalent of water flowing down a hydraulic gradi-
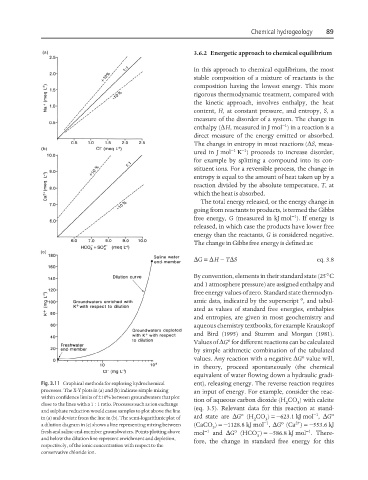
Fig. 3.11 Graphical methods for exploring hydrochemical ent), releasing energy. The reverse reaction requires
processes. The X-Y plots in (a) and (b) indicate simple mixing an input of energy. For example, consider the reac-
within confidence limits of ±10% between groundwaters that plot
tion of aqueous carbon dioxide (H CO ) with calcite
close to the lines with a 1 : 1 ratio. Processes such as ion exchange 2 3
(eq. 3.5). Relevant data for this reaction at stand-
and sulphate reduction would cause samples to plot above the line
o
−1
in (a) and deviate from the line in (b). The semi-logarithmic plot of ard state are ∆G (H CO ) =−623.1 kJ mol , ∆G o
2
3
2+
−1
o
a dilution diagram in (c) shows a line representing mixing between (CaCO ) =−1128.8 kJ mol , ∆G (Ca ) =−553.6 kJ
3
−
−1
fresh and saline end-member groundwaters. Points plotting above mol −1 and ∆G (HCO ) =−586.8 kJ mol . There-
o
and below the dilution line represent enrichment and depletion, 3
fore, the change in standard free energy for this
respectively, of the ionic concentration with respect to the
conservative chloride ion.